PluriPred : A webserver for predicting pluripotent proteins
|
PluriPred : A webserver for predicting pluripotent proteins |
HOME |
BROWSE |
ABOUT |
HELP |
TEAM |
|
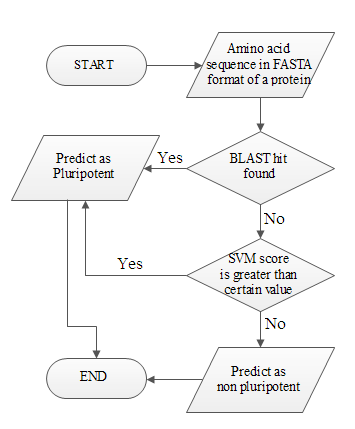
Pluripred is a web server for predicting whether a protein has an important role in pluripotency or not from amino acid sequence of the proteins. Datasets Positive dataset : PluriNetwork is a manually curated protein-protein interaction pluripotent network containing 274 mouse genes/proteins, which has direct evidence in pluripotency. Out of those genes 270 genes' ids were matched and used as positive training dataset to train the SVM model as well as making database for BLAST search. Negative dataset : Around 2785 genes were randomly selected from Uniprot for mouse genome those were not annotated in gene ontology with the terms, such as growth (GO:0040007, level 1), developmental process (GO: 0032502, level 1), cell proliferation (GO:0008283, level 1), cell differentiation (GO:0030154, level 4). CD-Hit was used for removing redundant genes with similar type of amino acid sequences. We selected sequence identity cut-off as 0.3 which represent the threshold of similarity between FASTA sequence clusters of proteins/genes and took the representative of the clusters. After that we manually deleted a few genes which were closely related. Finally, 932 genes were found which were used as negative dataset. Blind datasets : To validate our optimal model, two blind data sets were chosen that were not used in 5 fold cross validation. The first blind set was from ESCAPE database, where genome wide RNAi screening was used for identifying pluripotent genes. The second blind set was from PluriNet, which is first human pluripotent network constructed from expression data. Swiss-Prot genes(Mouse and human) : Amino acid sequences in FASTA format of 16232 mouse genes/proteins and 20193 human genes/proteins were collected from UniProt which had UniprotKB/Swiss-Prot entry and perform optimal model on those genes for predicting novel pluripotent genes/proteins. Downloads :Positive training dataset Negative training dataset Blind set from ESCAPE database Blind set from PluriNet Methods Since Support Vector Machine(SVM) model gives higher sensitivity, but low positive predictive value and BLAST search gives lower sensitivity but high positive predictive value, we proposed a hybrid model of these two, taking advantage of both models. The flow chart of our proposed hybrid model is given in Flow chart 1.
Feature vectors For training of our models we used different combinations of features which include
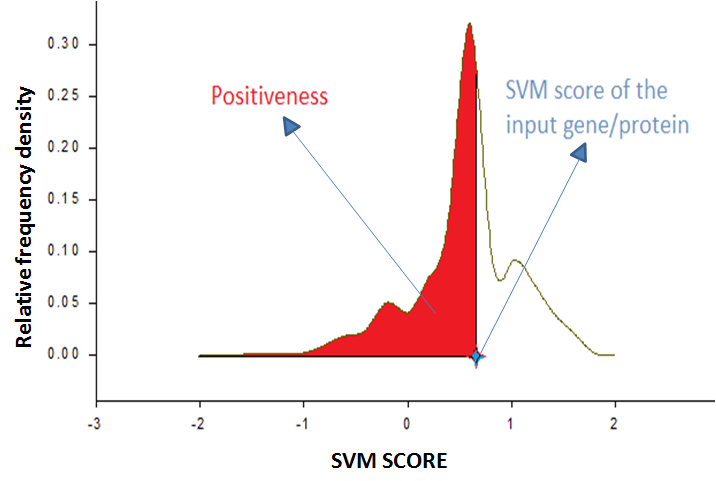
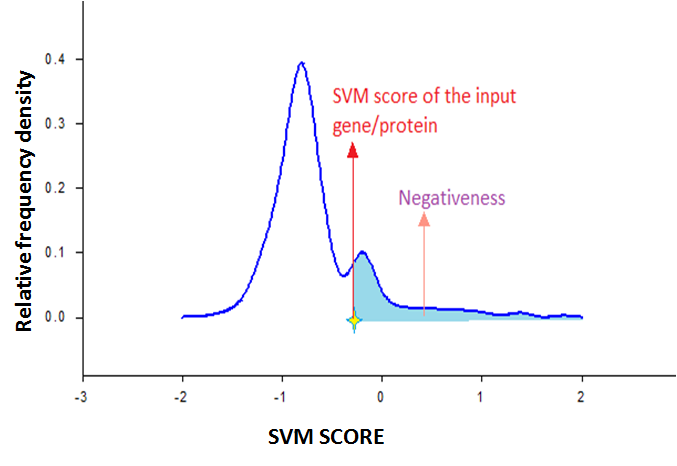
Confidence measure for the prediction Confidence measurement of prediction based on SVM score If we plot a histogram of a population with a bandwidth W, Fi is the frequency of within a particular bandwidth, and total size of the population is N then the density Di is Fi/(W*N) in this bandwidth.
If we multiply density with the bandwidth then we will get the relative frequency of the population for the bandwidth. The sum of all relative frequency is 1. So if we plot relative frequency density curve, then the area under the curve will be 1, and it represents the probability density function of the population.
In our model if we plot relative frequency plot of the positive training dataset as well as negative training dataset, it will give the probability density functions of the positive training dataset and negative training dataset respectively. From there we can calculate the chance of protein whether the protein is important or not for pluripotentcy from their SVM score that we denote in the terms of positiveness and negativeness.
Confidence measurement of prediction based on E-value of BLAST search The confidence of a protein to be pluripotent is also calculated in term of p-value from BLAST search. If minimum E-value of a protein is E with respect to the database of positive training dataset, then p-value is calculate by the equation is P = 1 – eE. Results 5 fold cross validation results : The results of different models are given in the Table 1. Table 1 : Performance compression of different models.
Blind set results : We validated our optimal model by pluripotent mouse proteins from ESCAPE database and also by human genes/proteins from PluriNet. Performance in unkown set from all Swiss-Prot proteins(Mouse and Human) : We evaluated all Swiss-Prot proteins of mouse and human by our proposed prediction model. For mouse we got 233 novel core pluripotent proteins and 323 novel extended pluripotent proteins. For human model, we got 167 novel core pluripotent and 385 extended pluripotent proteins.
Conclusion : This is an organism independent prediction server where it predicts pluripotent proteins/genes with their confidence level to be pluripotent protein/gene. So it can help the biological community by helping not only pluripotent stem cell research, but also in developmental biology, stem cell research and cancer research. |
|
© Bose Institute, Kolkata, 2015. All rights reserved. For any queries regarding PluriPred, contact Dr. Sudipto Saha at ssaha4@jcbose.ac.in |